I am Hirofumi Tsuruta, in charge of research and development on machine learning at COGNANO. We recently published a press release titled "A research paper from COGNANO with co-authors from Google on a large-scale dataset of antigen-antibody interactions was accepted by NeurIPS 2023". In this paper, we proposed a unique and promising methodology for generating a high-quality and large-scale dataset for drug discovery from alpacas. In this blog, we would like to focus on the important points of our NeurIPS accepted paper from the perspective of machine learning (ML).
Title | AVIDa-hIL6: A Large-Scale VHH Dataset Produced from an Immunized Alpaca for Predicting Antigen-Antibody Interactions |
Authors | Hirofumi Tsuruta, Hiroyuki Yamazaki, Ryota Maeda, Ryotaro Tamura, Jennifer N. Wei, Zelda Mariet, Poomarin Phloyphisut, Hidetoshi Shimokawa, Joseph R. Ledsam, Lucy Colwell, Akihiro Imura |
URL |
Antibody Drug Discovery and AI
Antibodies are proteins that play an essential role in the immune system. When antigens such as viruses and bacteria invade the body, the immune system protects the body by producing large numbers of antibodies that bind to the antigens to inhibit their function or mark them for removal. Antibodies have become an important class of therapeutic agents to treat human diseases because of their high target specificity and binding affinity. An essential step in therapeutic antibody discovery is the identification of specific interactions between antibody candidates and target antigens, called antigen-antibody interactions. This step has traditionally relied heavily on expensive, time-consuming experiments.
To accelerate therapeutic antibody discovery, computational methods, especially machine learning, have attracted interest for predicting antigen-antibody interactions. For example, Huang et al. [1] proposed AbAgIntPre, a deep learning-assisted prediction method that was trained using only the amino acid sequences in two public databases [2, 3]. Despite these promising developments, the publicly available datasets in existing works have notable limitations, such as small sizes and the lack of non-binding samples and exact amino acid sequences. For this reason, progress in therapeutic antibody discovery has lagged behind progress in other areas of drug discovery.
Data Generation Using Alpaca's Immune System
We proposed a novel method for generating a high-quality, large-scale dataset of antigen-antibody interactions using the immune system of a live alpaca and published the generated dataset, AVIDa-hIL6, on the following website. The dataset is released under a CC BY-NC 4.0 license.

The following figure briefly illustrates the key points on how to generate our dataset.
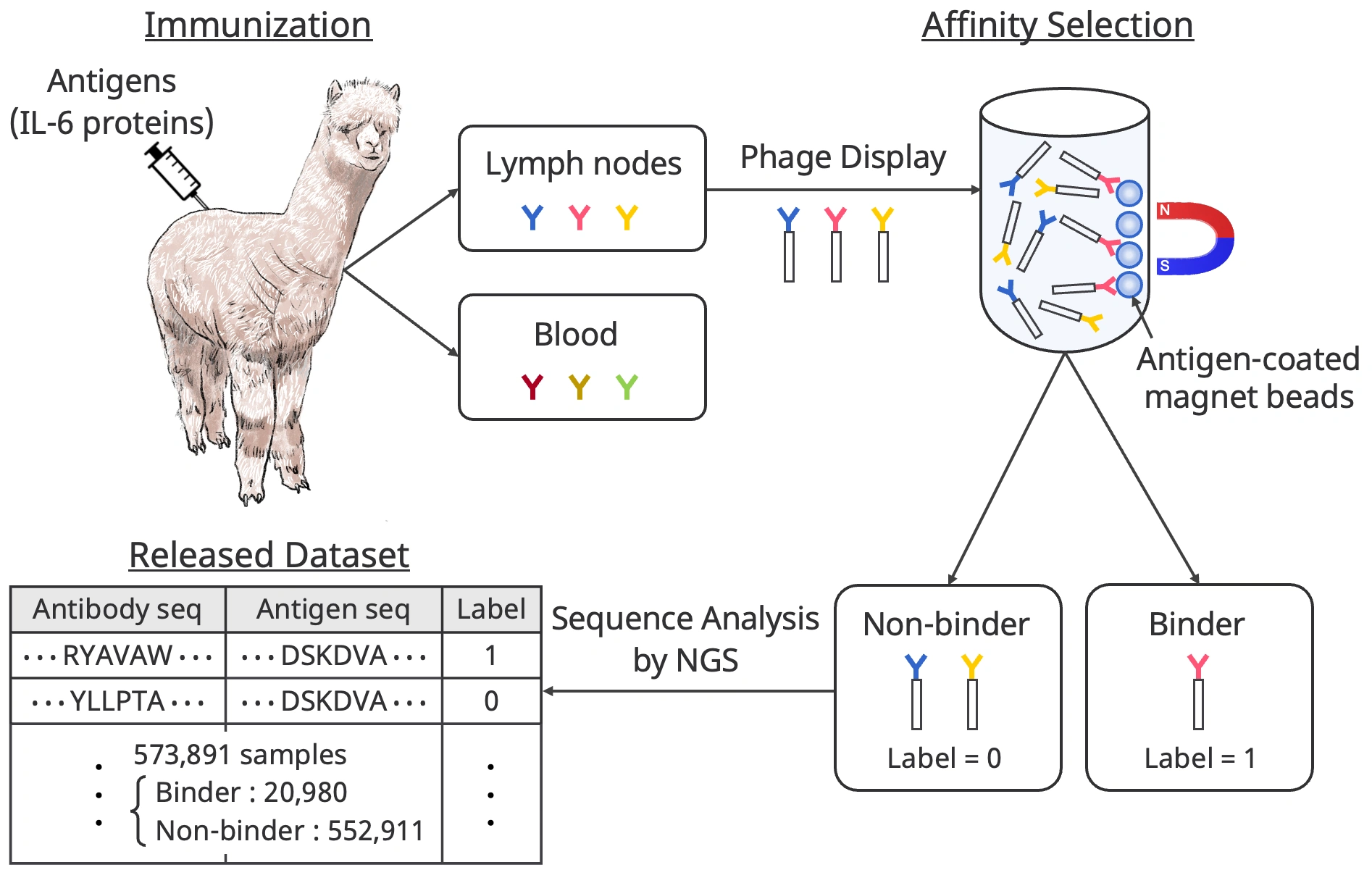
First, we immunized an alpaca with the human interleukin-6 (IL-6) protein that we used as an antigen. IL-6 is a relatively small protein, a simply structured, well-characterized cytokine that exists as a monomer in the body and is associated with many inflammatory diseases and cancers. After immunization, lymph nodes and blood samples were collected from the alpaca’s body. These samples contain a mixture of antibodies that bind to the IL-6 protein produced by the alpaca's immune response and ineffective antibodies that were originally in the alpaca's body. Thus, affinity selection, called biopanning, is used to distinguish between antibodies that bind to IL-6 protein (Binder) and those that do not (Non-binder). Then, the amino acid sequences of these antibodies were identified using next-generation sequencing (NGS).
The resulting dataset contains 573,891 data samples, including 20,980 binding pairs, with information on the amino acid sequence of the antigen (IL-6 protein), the amino acid sequence of the antibody, and the binary label of whether the antigen/antibody binds or not. To our knowledge, AVIDa-hIL6 is the largest public dataset for predicting antigen-antibody interactions.
Why Alpaca?
The main reason we use alpacas in our research is that alpacas have special antibodies that can efficiently identify amino acid sequences.
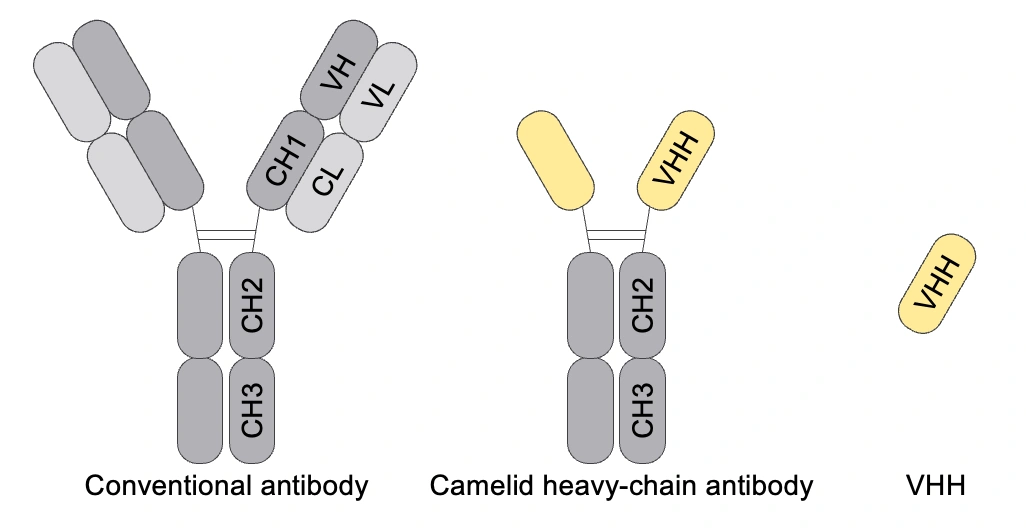
Conventional antibodies in humans, mice, and other animals consist of two heavy chains and two light chains. On the other hand, camelids such as alpacas and llamas have antibodies consisting only of heavy chains. Furthermore, the variable region of the heavy-chain antibodies is called VHH (or Nanobody), which is the smallest functional unit of heavy-chain antibodies. This simple structure of VHH enables much easier identification of full-length amino acid sequences by NGS than conventional antibodies.
Effects of Mutations
As the COVID-19 pandemic has shown, viruses continuously evolve through mutation to evade the immune system. Because emerging mutations involving amino acid substitutions can lead to profound changes in antibody binding, prediction of their effects is critical in the development of therapeutic antibodies.
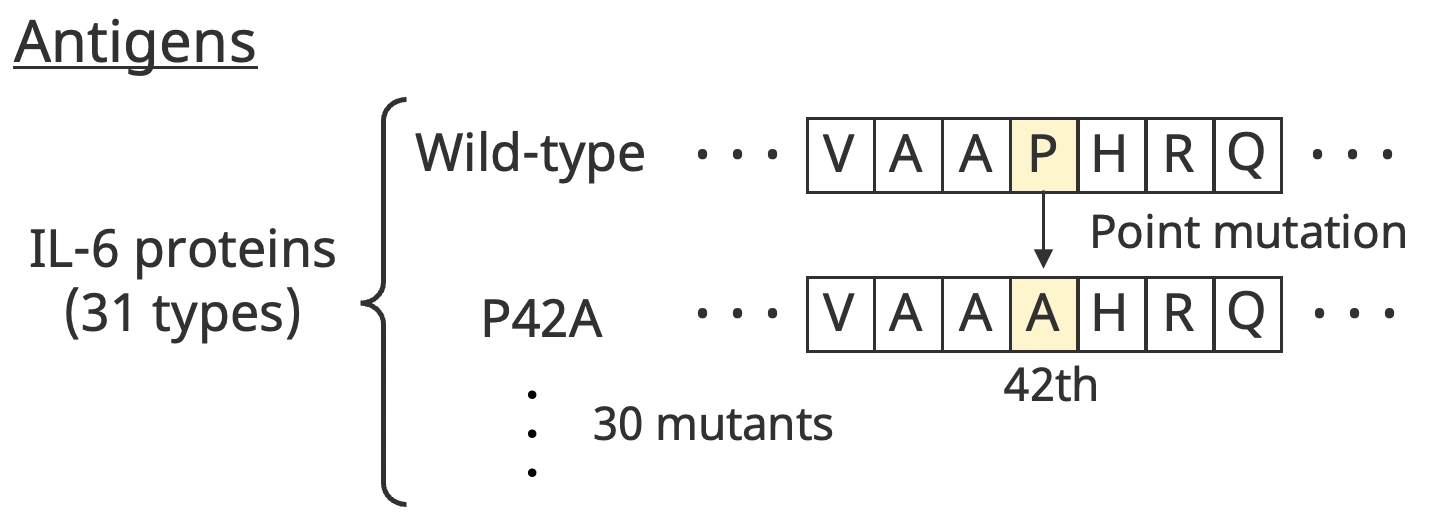
AVIDa-hIL6 contains information on the interaction of diverse VHHs with 30 different mutants produced by artificial point mutations, in addition to the wild-type IL-6 protein. For example, a mutant is denoted P42A, which means that an amino acid in the wild type is substituted from proline to alanine at position 42, as shown below.
I would like to explain the characteristics of our dataset in more detail using the following two figures.

The left figure shows the number of samples for each antigen type. Although the number of samples varied for each IL-6 protein type, we successfully generated at least 10,000 samples for the wild type and 30 different mutants. Furthermore, at least 250 binder VHH sequences existed for each IL-6 protein type.
The right figure shows the visualization of the labels for pairs of 100 VHH sequences and each antigen type. Each cell represents a unique VHH-antigen pair. Interestingly, these samples have valuable information on which mutations enhance or inhibit antibody binding, which should be strongly associated with the IL-6 protein's binding site.
Benchmarks with AVIDa-hIL6
In the paper, we reported benchmark results for predicting antigen-antibody interactions using several ML models.
Benchmark Task
A typical use of AVIDa-hIL6 is to solve a binary classification problem using the amino acid sequences of antigens and antibodies as input and binary labels as output. By leveraging information on the binding of diverse antibodies to antigen mutants, we defined a benchmark task to assess the model performance in capturing the impact of antigen mutations on antibody binding. Our experimental scenario assumes that antigen mutants emerge one after another to evade the immune system, as in the COVID-19 pandemic. In such a scenario, we evaluated the model's performance in predicting antibody candidates that will bind to future emerging mutants according to the binding information of antigens that have already been observed.
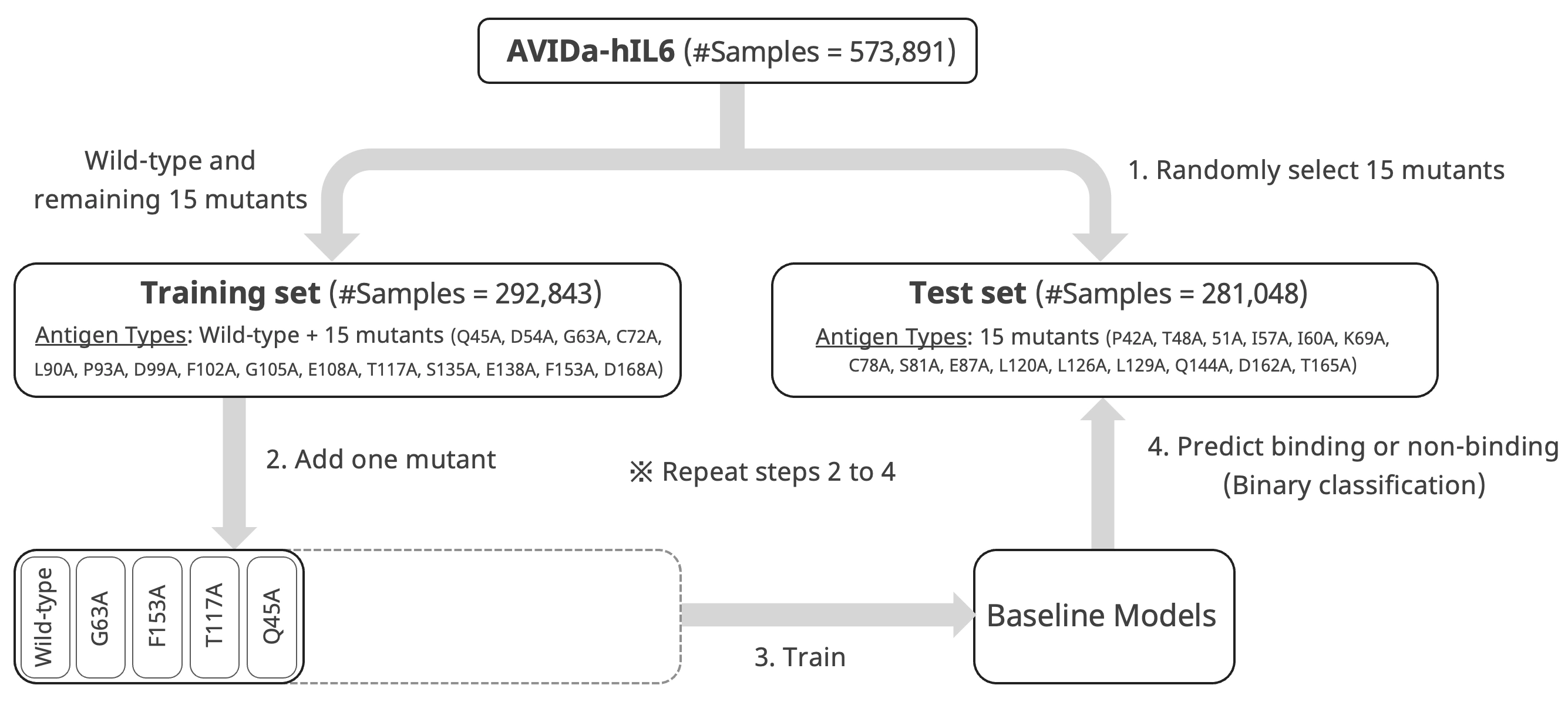
- We randomly selected 15 mutants and reserved the data samples for those mutants as a test set. The remaining 15 mutants and the wild type were reserved for model training.
- We randomly selected antigen type from the training set (the first is the wild type) and added it to the model training dataset.
- We trained the baseline models using a training dataset.
- We evaluated each model's predictive performance for the test set.
By repeating steps 2 to 4, we tracked the model's predictive performance for unknown mutants contained only in the test set.
Baseline Models
We adopted three neural network-based models and one classical machine learning model as baselines.
- AbAgIntPre [1] is a state-of-the-art model designed for antigen-antibody interactions based on amino acid sequences.
- PIPR [4] is a residual recurrent convolutional neural network for protein-protein interaction prediction.
- Multi-Layer Perceptron (MLP) with one hidden layer of 512 neurons was used as a simpler neural network-based model than the above two models.
- Logistic Regression (LR) was used as a classical machine learning model that is commonly used for binary classification tasks.
Results
The following figures show precision, recall, and F1-score as a function of the number of IL-6 protein types used for model training.
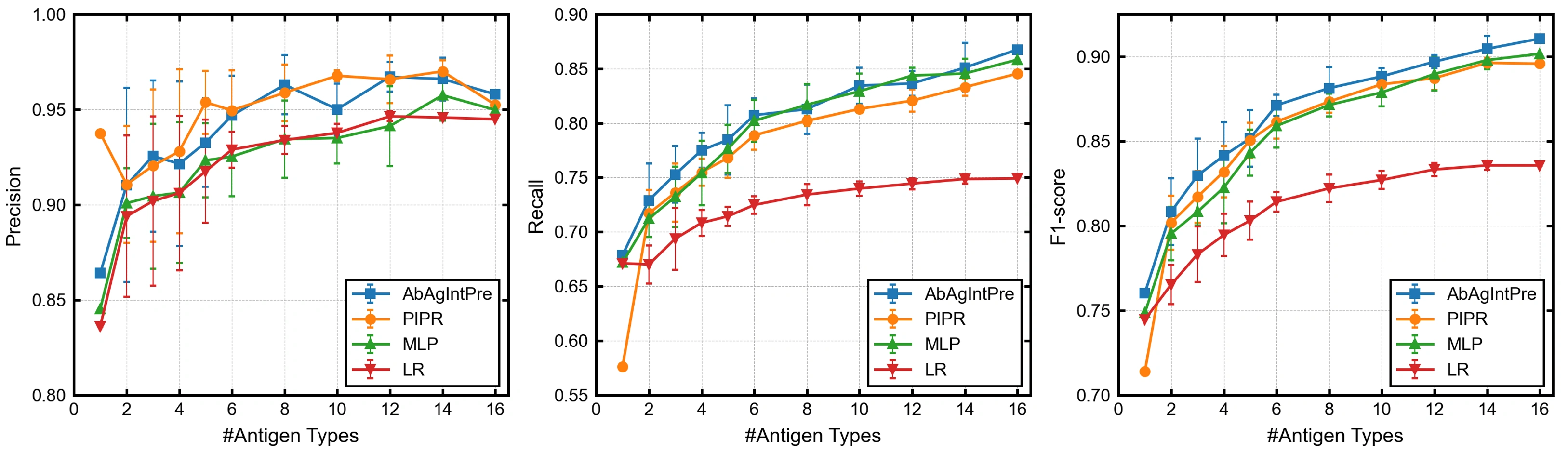
Here, we focus on recall, namely the accuracy with which the model could predict binding antibodies. When the number of antigens was 1---that is, when only the wild-type IL-6 protein was used for training---the recalls of AbAgIntPre, PIPR, MLP, and LR were 67.9, 57.6, 67.2, and 67.1 %, respectively. These results indicate that the models failed to predict over 30 % of the effective VHHs that bound to mutants in the test set. All the metrics improved as the number of IL-6 protein types used for training increased. However, after adding 15 mutants for training, the recalls were still only about 85 %, even for the three neural-network-based models.
For drug discovery applications, the construction of a generalized model for unknown mutations from as little antigen-binding information as possible is ideal, because the number of possible mutations in antigens is tremendously large. AVIDa-hIL6 differs significantly from the existing datasets used for training by AbAgIntPre and PIPR because it includes cases in which changes of a few amino acids enhance or inhibit antibody binding. Hence, these results indicate the need for research on model architectures that are dedicated to predicting antibody binding to antigen mutants, and AVIDa-hIL6 will be a useful benchmark for evaluating such models.
COGNANO's Challenge
The main limitation of AVIDa-hIL6 is the lack of antigen diversity: specifically, AVIDa-hIL6 only has the IL-6 protein as an antigen. These limitations lead to the narrow applicability of a model trained on AVIDa-hIL6. An essential approach to overcome this limitation will be to accumulate labeled data for a wider variety of antigens and their mutants. Because our data generation method is applicable to any target antigen, it can be a fundamental technology for establishing a more comprehensive database of antigen-antibody interactions. In fact, we used the same approach to generate a dataset for SARS-CoV-2 variants and successfully found effective antibodies [5]. In the future, we plan to generate and release datasets for various antigens, which should be more practical for building models to predict antigen-antibody interactions.

Closing thoughts
The released our dataset can be downloaded and used by anyone from this website. If you are interested, please give it a try! Thank you for taking the time to read our blog post.

References
[1] Huang, Y., Zhang, Z., Zhou, Y.: AbAgIntPre: A deep learning method for predicting antibody-antigen interactions based on sequence information. Frontiers in Immunology 13 (2022)
[2] Dunbar, J., Krawczyk, K., Leem, J., Baker, T., Fuchs, A., Georges, G., Shi, J., Deane, C.M.: SAbDab: the structural antibody database. Nucleic acids research 42(D1), D1140–D1146 (2014)
[3] Raybould, M.I., Kovaltsuk, A., Marks, C., Deane, C.M.: CoV-AbDab: the coronavirus antibody database. Bioinformatics 37(5), 734–735 (2021)
[4] Chen, M., Ju, C.J.T., Zhou, G., Chen, X., Zhang, T., Chang, K.W., Zaniolo, C., Wang, W.: Multifaceted protein–protein interaction prediction based on Siamese residual RCNN. Bioinformatics 35(14), i305–i314 (2019)
[5] Maeda, R., Fujita, J., Konishi, Y., Kazuma, Y., Yamazaki, H., Anzai, I., Watanabe, T., Yamaguchi, K., Kasai, K., Nagata, K., et al.: A panel of nanobodies recognizing conserved hidden clefts of all SARS-CoV-2 spike variants including Omicron. Communications Biology 5, 669 (2022)